Epigenetics is a word that has had a variety of meanings historically, and it's sometimes unclearly employed, even by the user. But these days, when people talk about epigenetics they generally mean the chemical modification of DNA sequence in a way that does not change the sequence itself but affects the expression of genes in or near the modified DNA region--that is why it is called 'epi' genetic. Such chemical modifications affect whether or not the cell uses a particular gene (only a subset of all genes is used in any particular cell, but that subset changes depending on the cell's local environment at any given time). That is, epigenetic changes essentially are regulatory; the epigenetically modified DNA sequences are not mutations of the coding of the structure of a particular protein (or a directly functional RNA), just how or when or how intensely it is used by the cell. Likewise, epigenetic modification doesn't change the affected sequence itself, but affects whether regulatory proteins can bind there to cause a nearby gene to be expressed, that is, transcribed into RNA. The phenomenon of such DNA 'marking' itself isn't controversial, and a few of the means by which it happens in a cell are known. Indeed, unlike mutations in the sequence itself, the marking is easily erased and there are known mechanisms that do that.
However, reports that epigenetic marking can be inherited are quite legitimately controversial. There are a few reasons for this.
How can local gene usage be inherited?
Cells respond to their environment--to extra-cellular conditions--via cell-surface receptors or other similar means. If they don't have receptors for a signal floating by, they can't detect or respond to that signal. But cells that do detect a signal change whether they start or stop using a particular set of genes. That's how complex multicellular, multi-organ system organisms become differentiated, as well as to respond to environmental conditions. Most examples of epigenetic inheritance relate to experience that affects particular types of cells, though many 'housekeeping' genes, genes that carry on basic metabolism, are used by all cells, and any environmental change could in principle induce all cells to change their gene usage.
Unless there is subsequent environmentally-induced change, once modified, when they divide, cells transmit their particular expression state to their daughter cells. If an epigenetic modification causes a cell to respond to a particular environmental signal by turning on the expression of a particular gene, that 'use it!' state would be passed on when the cell divides to produce other cells in its lineage, unless or until another modification occurred to reverse the original change. Thus, if some particular cell, say a lung cell, is induced by some environmental factor like a nutrient to express some set of lung-related genes, the effect is local, specific to lung cells. How that works is complex but some of the mechanisms are known. However, they have to do with how chromosomes specifically in lung cells are packaged; that is a local fact. For example, it need not also affect nerve or vessel or skin or stomach cells. Again, that is because in a differentiated organism different tissues are separated from each other so they can be different.
This raises a serious problem: Local effects on gene expression will be passed on to daughter cells in that tissue, but this is not the same as transmitting the effect to the next generation of organisms. Intergenerational transmission requires that the modification also be made in germline--sperm or egg cells--because the offspring organism starts out life just as a single fertilized egg (which has no lung cells!). Germline cells generally need to have genes switched on (or off) to enable them to make a new organism from scratch, from that single fertilized egg cell. Some temporary change that was important to the embryo's future lung cells would not likely be appropriate for the development of those cells in the first place during embryogenesis. So it is no surprise that there are active mechanisms to strip off epigenetic changes in germ cells' DNA, to reprogram those cells' gene usage to prepare them for their embryonic duties, this is done by erasing and re-setting DNA modification in the sperm or egg cell. If the embryo's lungs, when they eventually have them, need to modify what they due based on the air their exposed to, then new epigenetic changes will occur. Thus, the process of erasing and reprogramming removes those changes. Some bits of the genome are protected from this but it is not automatically true that even environmentally induced changes in housekeeping gene usage will be transmitted.
It was first systematically shown by Weismann in the 19th century and has been a theoretical bulwark against the idea of Lamarckian inheritance, that at least in most animals, somatic (body) and germline cells are separated, independent lineages isolated from each other (the situation is different in many or most plants). That means that for epigenetic changes to become heritable--and hence affect evolution--modifications to particular body cells would have to be applied to germline cells and not be erased before fertilization.
Without some clear mechanism, there is no reason that future sperm or egg cells will even 'know' about, much less respond to, the signal that induces change in the lung or nerve or stomach cells. So for epigenetic change to be inherited, there is the serious question of how the genomes in germline cells are specifically modified by signals that affect nerve or lung, etc. If a lung cell alters its use of gene X related to how lungs work, when it detects some (say) pollutant in the air, how does that specific change also get imposed on the germ line? Explanations that have been suggested so far are mainly not very convincing. That's why most reports of inherited epigenetic modification are properly received with skepticism.
Still, many investigators are seriously interested in epigenetic changes, especially when or if they are inherited, for a few reasons. This sort of inheritance, which modifies DNA usage differently among a person's many different localized tissues, threatens the degree to which traits can be predicted from a person's DNA sequence alone (obtained, for example, from a blood sample), and among other things that threatens realization of the promise of 'precision' genome-based medicine. Secondly, accurate assessment of epigenetic effects could lead to a better understanding of important environmental exposures and/or what to do about them, so that newborns are not doomed by their parents' habits to live with pre-set epigenetic traits that they now cannot prevent. And the least legitimate reason, but one important in the real world of today is that is a lucrative and sexy new finding that can be made to seem a melodramatic 'transformative' shift in our understanding of life.
An important criterion for claims of true epigenetic inheritance is that they must pass through at least to a 3d generation without the presence of the environmentally causal trigger. That is, transgenerational transmission is evidence that the genome is in fact preserving the change rather than just each new individual learning it from environmental experience (such as in utero). While there have been various generally convincing reports of true transgenerational inheritance in some species like the simple nematode (C. elegans) or plants, this hasn't clearly been shown in mammals (or humans), even if one or even two generational inheritance, usually through the maternal line, has been found.
Most of the literature consists of curious reports or claims of epigenetic inheritance, reviews of the germline erasure process and what areas of germline DNA could perhaps escape erasure of epigenetic marking, and some examples that seem to be truly transgenerational. At present, the excitement seems generally far exceeding the reality. But since epigenetics is potentially quite important, and the methods for understanding it rather new, it is being given serious attention.
A paper by Bohacek and Mansuy (November 2015 Nature Reviews Genetics), reviews what is known about the degree to which epigenetic 'marking' is inherited. This is a very good, measured paper that in our reading of it makes it clear that claims of non-trivial multi-generational DNA modification effects still need careful documentation. But if life-experience by parents can affect their offsprings' traits in substantial ways related to the offsprings' future life experience, even if they are not exposed to the risk factors that set their parents' genome usage patterns, then if we could understand how this works perhaps such modifications would not be destiny, and means of prevention or control could be developed if the phenomenon were to be better understood.
Gene usage isn't the same thing as gene structure
Epigenetic inheritance can also affect ideas about how evolution works, if they really have long-term (many generational) effects. The suggestion is now routinely being made that the phenotypic effects of epigenetics we are seeing introduces a Lamarckian view of evolution that may, after all, have to be melded with our Darwinian theory (e.g., see Skinner, MK, Gen Biol Evol. 7: 1296-1302, 2015). But the idea that this is a genuine revival of Lamarckism is still treated with sneering. Should it be?
We have written a 2015 series of posts about Lamarckian ideas. Lamarck was interested in the evolution of adaptive traits, like flying or ocean-living mammals, not just some specific minor traits. He had some non-starter ideas, but so did Darwin and they had far less knowledge than we do! So one can't defend his theory per se for various reasons. Still, it's worth thinking about rather than just sneering at Lamarck. That's for tomorrow's post.....
prediction etiketine sahip kayıtlar gösteriliyor. Tüm kayıtları göster
prediction etiketine sahip kayıtlar gösteriliyor. Tüm kayıtları göster
Unknowns, yes, but are there unknowables in biology?
The old Rumsfeld jokes about the knowns and unknowns are pretty stale by now, so we won't really indulge in beating that dead horse. But in fact his statement made a lot of sense. There are things we think we know (like our age), things we think we don't know but might know (like whether there will be a new message in our inbox when we sign onto email), and things we don't know but don't know we don't know (such as how many undiscovered marine species there are). Rumsfeld is the subject of ridicule not for this pronouncement per se (at least to those who think about it), because it is actually reasonable, but for other things that he is said to have done or said (or failed to say) in regard to American politics.
The unknowns may be problems, but they are not Big problems. What we don't know but might know are at least within the realm of learning. We may eventually stumble across facts we don't know but don't yet even know are there. The job of science is to learn what we know we don't know and even to discover what we don't yet know that we don't know. We think there is nothing 'inside' an electron or photon, but there may be if we some day realize that possibility. Then the guts of a photon will become a known unknown.
However, there's another, even more problematic, one may say truly problematic kind of mystery: things that are actually unknowable. They present a Really Big problem. For example, based on our understanding of the current understanding of cosmology, there are parts of the universe that are so far away that energy (light etc.) from them simply has not, and can never, reach us. We know that the details of this part of space are literally unknowable, but because we have reasonably rigorous physical theory we think we can at least reliably extrapolate from what we can see to the general contents (density of matter and galaxies etc.) of what we know must exist but cannot see. That is, it's literally unknowable but theoretically known.
However, things like whether life exists out there are in principle unknowable. But at least we know very specifically why that is so. In the future, most of what we can see in the sky today is, according to current cosmological theories, going to become invisible as the universe expands so that the light from these visible but distant parts will no longer be able to reach us. If there are any living descendants, they will know what was there to see and its dynamics and we will at least be able to make reasonable extrapolations of what it's like out there even though it can no longer be seen.
There are also 'multiverse' theories of various sorts (a book discussing these ideas is Our Mathematical Universe, by Mark Tegmark). At present, the various sorts of parallel universes are simply inaccessible, even in principle, so we can't really know anything about them (or, perhaps, even whether they exist). Not only is electromagnetic radiation not able to reach us so we can't observe, even indirectly, what was going on when that light was emitted from these objects, but our universe is self-contained relative to these other universes (if they exist).
Again, all of this is because of the kind of rigorous theory that we have, and the belief that if that theory is wrong, there is at least a correct theory to be discovered--Nature does work by fixed 'laws', and while our current understanding may have flaws the regularities we are finding are not imaginary even if they are approximations to something deeper (but comparably regular). In that sense, the theory we have tells us quite a lot about what seems likely to be the case even if unobserved. It was on such a basis that the Higgs boson was discovered (assuming the inferences from the LHC experiments are correct).
What about biology?
Biology has been rather incredibly successful in the last century and more. The discoveries of evolution and genetics are as great as those in any other science. But there remain plenty of unknowns about biological evolution and its genomic basis that are far deeper than questions about undiscovered species. We know that these things are unknown, but we presume they are knowable and will be understood some day.
One example is the way that homologous chromosomes (one inherited each of a person's parents) line up with each other in the first stage of meiosis (formation of sperm and egg cells). How do they find each other? We know they do line up when sex cells are produced, and there are some hypotheses and bits of relevant information about the process, but we're aware of the fact that we don't yet really know how it works.
Chromosomes also are arranged in a very different 3-dimensional way during the normal life of every cell. They form a spaghetti-like ball in the nucleus, with different parts of our 23 pairs of chromosomes very near to each other. This 'chromosome conformation', the specific spaghetti ball, shown schematically in the figure, varies among cell types, and even within a cell as it does different things. The reason seems to be at least in part that the juxtaposed bits of chromosomes contain DNA that is being transcribed (such as into messenger RNA to be translated into protein) in that particular cell under its particular circumstances.
It is easy to discuss what we don't know in evolution and genetics and we do that a lot here on MT. Often we critique current practice for claiming to know far more than is actually known, or, equally seriously, making promises to the supporting public that suggest we know things that in truth (and in private) we know very well that we don't know. In fact, we even know why some things that we promise are either unknown or known not to be correct (for example, causation of biological and behavioral traits is far more complex than is widely claimed).
There are pragmatic reasons why our current system of science does this, which we and many others have often discussed, but here we want to ask a different sort of question: Are there things in biology that are unknowable, even in principle, and if so how do we know that? The answer at least in part is 'yes', though that fact is routinely conveniently ignored.
Biological causation involves genetic and environmental factors. That is clearly known, in part because DNA is largely an inert molecule so any given bit of DNA 'does' something only in a particular context in the cell and related to whatever external factors affect the cell. But we know that the future environmental exposures are unknown, and we know that they are unknowable. What we will eat or do cannot be predicted even in principle, and indeed will be affected by what science learns but hasn't yet learned (if we find that some dietary factor is harmful, we will stop eating it and eat something else). There is no way to predict such knowledge or the response to it.
What else may there be of this sort?
A human has hundreds of billions of cells, a number which changes and varies among and within each of us. Each cell has a slightly different genotype and is exposed to slightly different aspects of the physical environment as well. One thing we know that we cannot now know is the genotype and environment of every cell at every time. We can make some statistical approximations, based on guessing about the countless unknowns of these details, but the numbers of variables will exceed that of stars on the universe and even in theory cannot be known with knowable precision.
Unlike much of physics, the use of statistical analytic techniques is inapt, also to an unknowable degree. We know that not all cells are identical observational units, for example, so that aggregate statistics that are used for decision-making (e.g., significance tests) are simply guesses or gross assumptions whose accuracy is unknowable. This is in principle because each cell, each individual is always changing. We might call these 'numerical unknowables', because they are a matter of practicality rather than theoretical limits about the phenomena themselves.
So are there theoretical aspects of biology that in some way we know are unknowable and not just unknown? We have no reason, based on current biological theory, to suspect the kinds of truly unknowables, analogous to cosmology's parallel universes. One can speculate about all sorts of things, such as parallel yous, and we can make up stories about how quantum uncertainty may affect us. But these are far from having the kind of cogency found in current physics.
Our lack of comparably rigorous theory relative to what physics and chemistry enjoy leaves open the possibility that life has its own knowably unknowables. If so, we would like at least to know what those limits may be, because much of biology relates to practical prediction (e.g., causes of disease). The state of knowledge in biology, no matter how advanced it has become, is still far from adequate to address the question of the levels of knowable things that may eventually be knowable, but also what the limits to knowability are. In a sense, unlike physics and cosmology, in biology we have no theory that tells us what we cannot know.
And unlike physics and cosmology, where some of these sorts of issues really are philosophical rather than of any practical relevance to daily life, we in biology have very strong reasons to want to know what we can know, and what we can promise....but perhaps also unlike physics, because people expect benefits from biological research, strong incentives not to acknowledge limits to our knowledge.
 |
| Explaining what we don't know is a problem! Source: Google images |
The unknowns may be problems, but they are not Big problems. What we don't know but might know are at least within the realm of learning. We may eventually stumble across facts we don't know but don't yet even know are there. The job of science is to learn what we know we don't know and even to discover what we don't yet know that we don't know. We think there is nothing 'inside' an electron or photon, but there may be if we some day realize that possibility. Then the guts of a photon will become a known unknown.
However, there's another, even more problematic, one may say truly problematic kind of mystery: things that are actually unknowable. They present a Really Big problem. For example, based on our understanding of the current understanding of cosmology, there are parts of the universe that are so far away that energy (light etc.) from them simply has not, and can never, reach us. We know that the details of this part of space are literally unknowable, but because we have reasonably rigorous physical theory we think we can at least reliably extrapolate from what we can see to the general contents (density of matter and galaxies etc.) of what we know must exist but cannot see. That is, it's literally unknowable but theoretically known.
However, things like whether life exists out there are in principle unknowable. But at least we know very specifically why that is so. In the future, most of what we can see in the sky today is, according to current cosmological theories, going to become invisible as the universe expands so that the light from these visible but distant parts will no longer be able to reach us. If there are any living descendants, they will know what was there to see and its dynamics and we will at least be able to make reasonable extrapolations of what it's like out there even though it can no longer be seen.
There are also 'multiverse' theories of various sorts (a book discussing these ideas is Our Mathematical Universe, by Mark Tegmark). At present, the various sorts of parallel universes are simply inaccessible, even in principle, so we can't really know anything about them (or, perhaps, even whether they exist). Not only is electromagnetic radiation not able to reach us so we can't observe, even indirectly, what was going on when that light was emitted from these objects, but our universe is self-contained relative to these other universes (if they exist).
Again, all of this is because of the kind of rigorous theory that we have, and the belief that if that theory is wrong, there is at least a correct theory to be discovered--Nature does work by fixed 'laws', and while our current understanding may have flaws the regularities we are finding are not imaginary even if they are approximations to something deeper (but comparably regular). In that sense, the theory we have tells us quite a lot about what seems likely to be the case even if unobserved. It was on such a basis that the Higgs boson was discovered (assuming the inferences from the LHC experiments are correct).
What about biology?
Biology has been rather incredibly successful in the last century and more. The discoveries of evolution and genetics are as great as those in any other science. But there remain plenty of unknowns about biological evolution and its genomic basis that are far deeper than questions about undiscovered species. We know that these things are unknown, but we presume they are knowable and will be understood some day.
One example is the way that homologous chromosomes (one inherited each of a person's parents) line up with each other in the first stage of meiosis (formation of sperm and egg cells). How do they find each other? We know they do line up when sex cells are produced, and there are some hypotheses and bits of relevant information about the process, but we're aware of the fact that we don't yet really know how it works.
 |
| Homologous chromosomes pair up...somehow. Wikimedia, public domain. |
Chromosomes also are arranged in a very different 3-dimensional way during the normal life of every cell. They form a spaghetti-like ball in the nucleus, with different parts of our 23 pairs of chromosomes very near to each other. This 'chromosome conformation', the specific spaghetti ball, shown schematically in the figure, varies among cell types, and even within a cell as it does different things. The reason seems to be at least in part that the juxtaposed bits of chromosomes contain DNA that is being transcribed (such as into messenger RNA to be translated into protein) in that particular cell under its particular circumstances.
 |
| Chromosomes arrange themselves systematically in the nucleus. Source: image by Cutkosky, Tarazi, and Lieberman-Aiden from Manoharan, BioTechniques, 2011 |
There are pragmatic reasons why our current system of science does this, which we and many others have often discussed, but here we want to ask a different sort of question: Are there things in biology that are unknowable, even in principle, and if so how do we know that? The answer at least in part is 'yes', though that fact is routinely conveniently ignored.
Biological causation involves genetic and environmental factors. That is clearly known, in part because DNA is largely an inert molecule so any given bit of DNA 'does' something only in a particular context in the cell and related to whatever external factors affect the cell. But we know that the future environmental exposures are unknown, and we know that they are unknowable. What we will eat or do cannot be predicted even in principle, and indeed will be affected by what science learns but hasn't yet learned (if we find that some dietary factor is harmful, we will stop eating it and eat something else). There is no way to predict such knowledge or the response to it.
What else may there be of this sort?
A human has hundreds of billions of cells, a number which changes and varies among and within each of us. Each cell has a slightly different genotype and is exposed to slightly different aspects of the physical environment as well. One thing we know that we cannot now know is the genotype and environment of every cell at every time. We can make some statistical approximations, based on guessing about the countless unknowns of these details, but the numbers of variables will exceed that of stars on the universe and even in theory cannot be known with knowable precision.
Unlike much of physics, the use of statistical analytic techniques is inapt, also to an unknowable degree. We know that not all cells are identical observational units, for example, so that aggregate statistics that are used for decision-making (e.g., significance tests) are simply guesses or gross assumptions whose accuracy is unknowable. This is in principle because each cell, each individual is always changing. We might call these 'numerical unknowables', because they are a matter of practicality rather than theoretical limits about the phenomena themselves.
So are there theoretical aspects of biology that in some way we know are unknowable and not just unknown? We have no reason, based on current biological theory, to suspect the kinds of truly unknowables, analogous to cosmology's parallel universes. One can speculate about all sorts of things, such as parallel yous, and we can make up stories about how quantum uncertainty may affect us. But these are far from having the kind of cogency found in current physics.
Our lack of comparably rigorous theory relative to what physics and chemistry enjoy leaves open the possibility that life has its own knowably unknowables. If so, we would like at least to know what those limits may be, because much of biology relates to practical prediction (e.g., causes of disease). The state of knowledge in biology, no matter how advanced it has become, is still far from adequate to address the question of the levels of knowable things that may eventually be knowable, but also what the limits to knowability are. In a sense, unlike physics and cosmology, in biology we have no theory that tells us what we cannot know.
And unlike physics and cosmology, where some of these sorts of issues really are philosophical rather than of any practical relevance to daily life, we in biology have very strong reasons to want to know what we can know, and what we can promise....but perhaps also unlike physics, because people expect benefits from biological research, strong incentives not to acknowledge limits to our knowledge.
Who should take statins? Is heart disease predictable?
Who should take statins.....besides everyone? I thought a lot about this when I was working on a lecture about predicting disease. The purpose of statins, of course, is to prevent atherosclerotic cardiovascular disease in people at risk (how well they do this is another issue). The challenge is to identify the people 'at risk'. I wrote about this in July, but I've been playing some more with the ideas and wanted to follow up.
Statins are a class of drug that, in theory, work by lowering LDL (low-denstity lipoprotein) levels. They do this by inhibiting HMG-CoA reductase, an enzyme that has a central role in the production of cholesterol in the liver. LDL, the so-called 'bad' cholesterol, isn't actually just cholesterol, but has been linked to risk of heart disease because, as a lipoprotein, its job is to transport cholesterol to and from cells. It is bound to cholesterol. What's measured when we have our blood drawn for a cholesterol test is LDL-C, the amount of cholesterol bound to LDL particles (LDL-C), as well as HDL-C, the 'good' cholesterol package, which transports LDL-C from cells, leading to lower blood cholesterol levels. Cholesterol makes plaque and plaque lines and hardens arteries, which occludes them and leads to stroke and heart attack. Lower the amount of LDL, and you lower the risk of arterial plaque deposits.
The connection between cholesterol and heart disease was first identified in the Framingham Study in the 1950's and 60's, and this lead directly to the search for drugs to lower cholesterol. Statins were developed in the 1970's and 80's, and after some fits and starts, began to be used in earnest in the late 1980's. Statins work by inhibiting the liver cells' synthesizing of new cholesterol, that is, cholesterol that isn't due taken in in the diet.
Akira Endo, one of the first scientists to look for cholesterol-lowering compounds, reviewed the history of statins in 2010. He described the many studies of the effects of these drugs, saying "The results in all these studies have been consistent: treatment with statins lowers plasma LDL levels by 25–35% and reduces the frequency of heart attacks by 25–30%" (Akira Endo, Proc Japan Acad, Series B, 2010).
A systematic review of the literature on the effectiveness of statins was published by the Cochrane Organization in 2012. The review reports, "Of 1000 people treated with a statin for five years, 18 would avoid a major CVD event which compares well with other treatments used for preventing cardiovascular disease." This suggests, of course, that 982 people took statins with no benefit, and perhaps some risk, as statins are associated with muscle pain, slightly increased risk of type 2 diabetes, liver damage, neurological effects, digestive problems, rash and flushing, and other effects. But more on this below.
So, who should take statins?
Until 2013, the recommendation was that anyone with a modest risk, as assessed by the Framingham Risk Calculator (I've read that that means from 6.5% to 10% 10-year risk) would likely be prescribed statins. The interesting thing, to me, about this risk calculator is that it's impossible to push the risk estimate past "greater than 30%", even at maximum allowable cholesterol, LDL, and systolic blood pressure, and being a smoker on blood pressure medication. Which means that there's a lot that this calculator can't tell us about our risk of CVD, based on the best risk factors known.
In 2013, the American Heart Association/American College of Cardiology revised their criteria for statins. Now, they are recommended for people who have had one CVD event in order to prevent another; for people with primary elevations of LDL-C greater than 190mg/dL; people 45-70 years old who have diabetes and LDL-C between 70 and 189mg/dL, and people 45-70 years old with LDL-C between 70 and 189mg/dL and estimated 10-year cardiovascular disease risk of 7.5% or higher.
The first three criteria are straightforward. If statins lower LDL, and lower LDL lowers risk of ASCVD (artherosclerotic cardiovascular disease), then taking them should be beneficial. But then we're back to a risk calculator again to estimate 10-year risk.
It has been revised. Now included are ethnicity (well, White, African American or other), and diabetic status (yes/no), and estimated lifetime risk. And, now it's possible to push 10-year risk up past 70%, which I discovered by playing around with the calculator a bit. Whether or not it's a more accurate predictor of a cardiovascular event is another question.
Here's the lowest risk I could come up with, 0.1% 10-year risk. The recommendations offered are not to prescribe statins.
Here's the highest risk I could force the calculator to estimate. Ten-year risk for a female with these risk factors is higher than for a male, but lifetime risk is lower. That seems strange, but ok, it must reflect association of risk factors including sex with disease at the population level.

Compared with the Framingham calculator, risk estimation seems to be getting more precise. Or at least bolder, with estimates up in the 70's. But is the new calculator actually better at predicting risk than the old one? A paper was recently published in JAMA addressing just this question ("Guideline-
Based Statin Eligibility, Coronary Artery Calcification, and Cardiovascular Events," Pursnani et al.) They identified 2435 people from the Framingham study who had never taken statins. Their medical history allowed the authors to determine that, based on the old guidelines, 14% would have been 'statin eligible' compared with 39%, based on the new 2013 guidelines.
Among those eligible by the old guidelines, 6.9% (24/348) developed CVD compared with 2.4% (50/2087) among noneligible participants (HR, 3.1; 95% CI, 1.9-5.0; P less than .001). Under the new guidelines, among those eligible for statins, 6.3% (59/941) developed incident CVD compared with only 1.0% (15/1494) among those not eligible (HR, 6.8; 95% CI, 3.8-11.9; P less than .001).
So, put a whole lot more people on statins, and you prevent an additional very small number of CVD events; 1.0% vs 2.4%. And, 93% of those ‘eligible’ for statins did not develop disease. Nor, of course, do statins prevent all disease. Actually, if everyone in the population were covered, statins would be preventing as many events as they could possibly prevent, but in a small minority of the population. That is, 90+% of people considered to be at 'high-risk' of disease don't go on to develop disease. Is it worth the side effects and cost to put so many more people on statins to prevent the 1.4% more CVD that these new guidelines are preventing? Well, heart disease is still the number one killer in rich countries, and 40+% of the population is currently taking statins, so a lot of people have decided that the benefits do outweigh the risks.
Another question, though, is more fundamental, and it concerns prediction. The calculator seems to now be predicting risk with some confidence. But, let's take a hypothetical person with a somewhat elevated risk. Her cholesterol is higher than the person above who's at lowest risk, but that's due to her HDL. Her systolic blood pressure is high at 180, which is apparently what bumps up her risk, but her 10-year risk is still not over 7.5% so the recommendation is not statins, but lifestyle and nutrition counseling. (Though, the definition of 'heart-healthy diet' keeps changing, so what to counsel this person with low risk seems a bit problematic, but ok.)
Now here's the same hypothetical person, but she's now a smoker, on medication to lower her blood pressure (and her b.p. is still high) and she has diabetes. Her 10-year risk of ASCVD jumps to 36.8%. This makes sense, given what we know about risk factors, right? The recommendation for her is high-intensity statins and lifestyle changes -- lose weight, do regular aerobic exercise, eat a heart-healthy diet, stop smoking (easy enough to say, so hard to do, which is another issue, of course, and the difficulty of changing all these behaviors is one reason that statins are so commonly prescribed).

But now I've lowered her total cholesterol by 70mg/dL, which is what statins ideally would do for her. Even so, the American College of Cardiology/American Heart Association recommendation is for 'high-intensity statin therapy' and lifestyle counseling. The calculator doesn't know this, but statins have already done everything they are likely to do for her.

So, let's add lifestyle changes. But, even when she quits smoking, her 10-year risk is 20%. So let's say we cure her diabetes -- even then, she's still at high enough risk (9%) that 'moderate to high-intensity statins' are recommended. I'm confused. I think even the calculator is confused. It seems there's a fuzzy area where statins are being recommended when what's left to do is, say, lower blood pressure, which statins won't do. This hypothetical woman probably needs to lower her weight to do that, and statins aren't going to help with that, either, but still they're recommended. Indeed, one of the criticisms of this risk calculator when it was released in 2013 was that it overestimates risk. Perhaps so, but it also seems to overestimate the benefit of statins.
Statins are a class of drug that, in theory, work by lowering LDL (low-denstity lipoprotein) levels. They do this by inhibiting HMG-CoA reductase, an enzyme that has a central role in the production of cholesterol in the liver. LDL, the so-called 'bad' cholesterol, isn't actually just cholesterol, but has been linked to risk of heart disease because, as a lipoprotein, its job is to transport cholesterol to and from cells. It is bound to cholesterol. What's measured when we have our blood drawn for a cholesterol test is LDL-C, the amount of cholesterol bound to LDL particles (LDL-C), as well as HDL-C, the 'good' cholesterol package, which transports LDL-C from cells, leading to lower blood cholesterol levels. Cholesterol makes plaque and plaque lines and hardens arteries, which occludes them and leads to stroke and heart attack. Lower the amount of LDL, and you lower the risk of arterial plaque deposits.
The connection between cholesterol and heart disease was first identified in the Framingham Study in the 1950's and 60's, and this lead directly to the search for drugs to lower cholesterol. Statins were developed in the 1970's and 80's, and after some fits and starts, began to be used in earnest in the late 1980's. Statins work by inhibiting the liver cells' synthesizing of new cholesterol, that is, cholesterol that isn't due taken in in the diet.
Akira Endo, one of the first scientists to look for cholesterol-lowering compounds, reviewed the history of statins in 2010. He described the many studies of the effects of these drugs, saying "The results in all these studies have been consistent: treatment with statins lowers plasma LDL levels by 25–35% and reduces the frequency of heart attacks by 25–30%" (Akira Endo, Proc Japan Acad, Series B, 2010).
A systematic review of the literature on the effectiveness of statins was published by the Cochrane Organization in 2012. The review reports, "Of 1000 people treated with a statin for five years, 18 would avoid a major CVD event which compares well with other treatments used for preventing cardiovascular disease." This suggests, of course, that 982 people took statins with no benefit, and perhaps some risk, as statins are associated with muscle pain, slightly increased risk of type 2 diabetes, liver damage, neurological effects, digestive problems, rash and flushing, and other effects. But more on this below.
So, who should take statins?
Until 2013, the recommendation was that anyone with a modest risk, as assessed by the Framingham Risk Calculator (I've read that that means from 6.5% to 10% 10-year risk) would likely be prescribed statins. The interesting thing, to me, about this risk calculator is that it's impossible to push the risk estimate past "greater than 30%", even at maximum allowable cholesterol, LDL, and systolic blood pressure, and being a smoker on blood pressure medication. Which means that there's a lot that this calculator can't tell us about our risk of CVD, based on the best risk factors known.
 |
| Framingham Risk Calculator |
In 2013, the American Heart Association/American College of Cardiology revised their criteria for statins. Now, they are recommended for people who have had one CVD event in order to prevent another; for people with primary elevations of LDL-C greater than 190mg/dL; people 45-70 years old who have diabetes and LDL-C between 70 and 189mg/dL, and people 45-70 years old with LDL-C between 70 and 189mg/dL and estimated 10-year cardiovascular disease risk of 7.5% or higher.
The first three criteria are straightforward. If statins lower LDL, and lower LDL lowers risk of ASCVD (artherosclerotic cardiovascular disease), then taking them should be beneficial. But then we're back to a risk calculator again to estimate 10-year risk.
 |
| ACC/AHA |
It has been revised. Now included are ethnicity (well, White, African American or other), and diabetic status (yes/no), and estimated lifetime risk. And, now it's possible to push 10-year risk up past 70%, which I discovered by playing around with the calculator a bit. Whether or not it's a more accurate predictor of a cardiovascular event is another question.
Here's the lowest risk I could come up with, 0.1% 10-year risk. The recommendations offered are not to prescribe statins.
 |
| Lowest 10-year risk |

Compared with the Framingham calculator, risk estimation seems to be getting more precise. Or at least bolder, with estimates up in the 70's. But is the new calculator actually better at predicting risk than the old one? A paper was recently published in JAMA addressing just this question ("Guideline-
Based Statin Eligibility, Coronary Artery Calcification, and Cardiovascular Events," Pursnani et al.) They identified 2435 people from the Framingham study who had never taken statins. Their medical history allowed the authors to determine that, based on the old guidelines, 14% would have been 'statin eligible' compared with 39%, based on the new 2013 guidelines.
Among those eligible by the old guidelines, 6.9% (24/348) developed CVD compared with 2.4% (50/2087) among noneligible participants (HR, 3.1; 95% CI, 1.9-5.0; P less than .001). Under the new guidelines, among those eligible for statins, 6.3% (59/941) developed incident CVD compared with only 1.0% (15/1494) among those not eligible (HR, 6.8; 95% CI, 3.8-11.9; P less than .001).
So, put a whole lot more people on statins, and you prevent an additional very small number of CVD events; 1.0% vs 2.4%. And, 93% of those ‘eligible’ for statins did not develop disease. Nor, of course, do statins prevent all disease. Actually, if everyone in the population were covered, statins would be preventing as many events as they could possibly prevent, but in a small minority of the population. That is, 90+% of people considered to be at 'high-risk' of disease don't go on to develop disease. Is it worth the side effects and cost to put so many more people on statins to prevent the 1.4% more CVD that these new guidelines are preventing? Well, heart disease is still the number one killer in rich countries, and 40+% of the population is currently taking statins, so a lot of people have decided that the benefits do outweigh the risks.
Another question, though, is more fundamental, and it concerns prediction. The calculator seems to now be predicting risk with some confidence. But, let's take a hypothetical person with a somewhat elevated risk. Her cholesterol is higher than the person above who's at lowest risk, but that's due to her HDL. Her systolic blood pressure is high at 180, which is apparently what bumps up her risk, but her 10-year risk is still not over 7.5% so the recommendation is not statins, but lifestyle and nutrition counseling. (Though, the definition of 'heart-healthy diet' keeps changing, so what to counsel this person with low risk seems a bit problematic, but ok.)
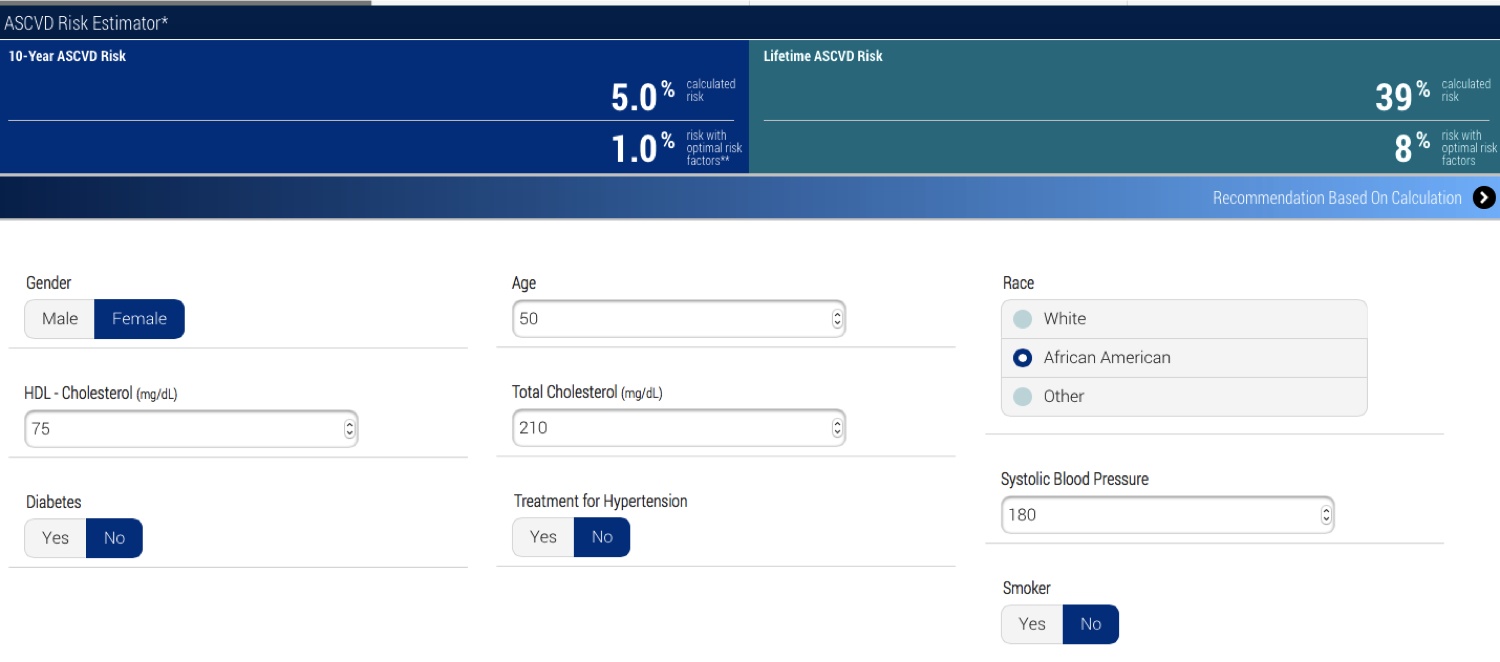 |
| Low enough risk that statins aren't advised. |
Now here's the same hypothetical person, but she's now a smoker, on medication to lower her blood pressure (and her b.p. is still high) and she has diabetes. Her 10-year risk of ASCVD jumps to 36.8%. This makes sense, given what we know about risk factors, right? The recommendation for her is high-intensity statins and lifestyle changes -- lose weight, do regular aerobic exercise, eat a heart-healthy diet, stop smoking (easy enough to say, so hard to do, which is another issue, of course, and the difficulty of changing all these behaviors is one reason that statins are so commonly prescribed).

But now I've lowered her total cholesterol by 70mg/dL, which is what statins ideally would do for her. Even so, the American College of Cardiology/American Heart Association recommendation is for 'high-intensity statin therapy' and lifestyle counseling. The calculator doesn't know this, but statins have already done everything they are likely to do for her.

So, let's add lifestyle changes. But, even when she quits smoking, her 10-year risk is 20%. So let's say we cure her diabetes -- even then, she's still at high enough risk (9%) that 'moderate to high-intensity statins' are recommended. I'm confused. I think even the calculator is confused. It seems there's a fuzzy area where statins are being recommended when what's left to do is, say, lower blood pressure, which statins won't do. This hypothetical woman probably needs to lower her weight to do that, and statins aren't going to help with that, either, but still they're recommended. Indeed, one of the criticisms of this risk calculator when it was released in 2013 was that it overestimates risk. Perhaps so, but it also seems to overestimate the benefit of statins.
Further, it seems there are a lot of type 1 errors here. That is, a lot of people are considered 'at-risk' who wouldn't actually develop cardiovascular disease. Risk of 7.5% means 7.5 of 100 people with a given, equal set of risk factors are expected to develop disease. That means that 92.5 would not. And that means that we have a pretty rough understanding of heart disease risk. The strongest risk factors we know -- smoking, high LDL-C, diabetes and hypertension -- can be expected to predict only a small fraction of events.
And that means that either something else is 'causing' cardiovascular disease in addition to these major known risk factors, or something is protecting people with these risk factors who don't go on to develop disease. Family history is a good or even the very best single predictor (why isn't it taken into account in these calculators?) which suggests that it's possible that genetic risk (or protection) is involved, but genome wide association studies haven't found genes with large effects. Of course, family history is highly conflated with environmental factors, too, so we shouldn't simply assume we need to look for genes when family history indicates risk. Anyway, it's unlikely that there are single genes responsible for ASCVD except in rare families, because that's the nature of complex diseases. Instead, many genes would be involved, but again as with most complex diseases, they would surely be interacting with environmental risk factors, and we don't yet know understand how to identify or really understand gene by environment interaction.
And then there's the truly wild card! All of these risks are based on the combinations of past exposures to measured lifestyle factors, but the mix of those and the rise of other new lifestyle factors, or the demise of past ones, means that the most fundamental of all predictors can itself not be predicted, not even in principle!
So, statins are a very broad brush, and a lot more people are being painted with them than in fact need to be. The problem is determining which people these are, but rather than zoom in with more precision, the updated calculator instead paints a whole lot more people with the brush. This isn't the calculator's fault. It's because understanding risk is difficult, ASCVD is a large and heterogeneous category, and prediction is very imprecise -- even for many 'simple' Mendelian disorders. If ASCVD were caused by a single gene, we'd say it had very low penetrance. And we'd want to understand the factors that affect its penetrance. That's the equivalent to where we are with cardiovascular disease.
I was interested to see that the 2013 ACC/AHA Guideline on the Assessment of Cardiovascular Risk says something that I have said so many times that I decided not to say it again in this post. But, I'm happy to see it elsewhere now. The guideline committee itself acknowledges the issue, so I'll let them explain the problem of assessing risk as their calculator does.
Finally, here's a meta-thought about all this. Ken and I were in Finland this month co-teaching a course, Logical Reasoning in Human Genetics, with colleagues, including Joe Terwilliger. Joe said multiple times, "We suck at finding candidate genes because we don't know anything about biology. We're infants learning to crawl." The same can be said about epidemiological risk factors for many complex diseases -- we suck at understanding the causes of these diseases, and thus we suck at prediction, because we don't really understand the biology.
And that means that either something else is 'causing' cardiovascular disease in addition to these major known risk factors, or something is protecting people with these risk factors who don't go on to develop disease. Family history is a good or even the very best single predictor (why isn't it taken into account in these calculators?) which suggests that it's possible that genetic risk (or protection) is involved, but genome wide association studies haven't found genes with large effects. Of course, family history is highly conflated with environmental factors, too, so we shouldn't simply assume we need to look for genes when family history indicates risk. Anyway, it's unlikely that there are single genes responsible for ASCVD except in rare families, because that's the nature of complex diseases. Instead, many genes would be involved, but again as with most complex diseases, they would surely be interacting with environmental risk factors, and we don't yet know understand how to identify or really understand gene by environment interaction.
And then there's the truly wild card! All of these risks are based on the combinations of past exposures to measured lifestyle factors, but the mix of those and the rise of other new lifestyle factors, or the demise of past ones, means that the most fundamental of all predictors can itself not be predicted, not even in principle!
So, statins are a very broad brush, and a lot more people are being painted with them than in fact need to be. The problem is determining which people these are, but rather than zoom in with more precision, the updated calculator instead paints a whole lot more people with the brush. This isn't the calculator's fault. It's because understanding risk is difficult, ASCVD is a large and heterogeneous category, and prediction is very imprecise -- even for many 'simple' Mendelian disorders. If ASCVD were caused by a single gene, we'd say it had very low penetrance. And we'd want to understand the factors that affect its penetrance. That's the equivalent to where we are with cardiovascular disease.
I was interested to see that the 2013 ACC/AHA Guideline on the Assessment of Cardiovascular Risk says something that I have said so many times that I decided not to say it again in this post. But, I'm happy to see it elsewhere now. The guideline committee itself acknowledges the issue, so I'll let them explain the problem of assessing risk as their calculator does.
By its nature, such an approach requires a platform for reliable quantitative estimation of absolute risk based on data from representative population samples. It is important to note that risk estimation is based on group averages, which are then applied to individual patients in practice. This process is admittedly imperfect; no one has 10% or 20% of a heart attack during a 10-year period. Individuals with the same estimated risk will either have or not have the event of interest, and only those patients who are destined to have an event can have their event prevented by therapy.It's the problem of using group data, which is all we've got, to make clinical decisions about individuals. It's the meta-analysis problem -- meta-analyses compile data from many individual studies to produce a single result that certainly reflects all the studies, because they were all included in the statistics, but it doesn't represent any of them with precision. Ultimately, it's the problem that these sorts of inferences must be based on statistical analysis of samples -- collections -- of individuals. We do not have an easy way around this, including the N of 1 studies currently being proposed.
Finally, here's a meta-thought about all this. Ken and I were in Finland this month co-teaching a course, Logical Reasoning in Human Genetics, with colleagues, including Joe Terwilliger. Joe said multiple times, "We suck at finding candidate genes because we don't know anything about biology. We're infants learning to crawl." The same can be said about epidemiological risk factors for many complex diseases -- we suck at understanding the causes of these diseases, and thus we suck at prediction, because we don't really understand the biology.
Kaydol:
Kayıtlar (Atom)
Rare Disease Day and the promises of personalized medicine
O ur daughter Ellen wrote the post that I republish below 3 years ago, and we've reposted it in commemoration of Rare Disease Day, Febru...
-
 Pakistan dizileri Hint dizilerinden farklı. Onlar gibi coşkulu olmuyor genelde. Bu yüzden yarım bıraktıklarım hayli fazla. Ama bu dizi ...
Pakistan dizileri Hint dizilerinden farklı. Onlar gibi coşkulu olmuyor genelde. Bu yüzden yarım bıraktıklarım hayli fazla. Ama bu dizi ... -
 O ur daughter Ellen wrote the post that I republish below 3 years ago, and we've reposted it in commemoration of Rare Disease Day, Febru...
O ur daughter Ellen wrote the post that I republish below 3 years ago, and we've reposted it in commemoration of Rare Disease Day, Febru... -
 W e discussed a Japanese pachinko machine in an earlier post , a pinball machine, as an example of the difference between randomness and det...
W e discussed a Japanese pachinko machine in an earlier post , a pinball machine, as an example of the difference between randomness and det...