I have a doctorate in Public Health which means that, unlike a 'real doctor', I was trained to think in terms of the health of populations, not of specific individuals. Public Health of course, when done appropriately, can have an enormous impact on the health of individuals, but in a very real way that's a side effect of gathering group information and instituting measures meant to affect a group. Clean water, fluoridated water, vaccinations, window screens, anti-smoking campaigns, and so much more are all public health measures targeting whole populations, without regard for the specific cavities or cases of cholera or lung cancer that the measure will actually prevent. This is because, of course, smoking doesn't make every smoker sick, just enough of them that aiming to convince whole populations not to smoke can have a large enough difference on population health that it's worth the cost and effort.
You've probably seen those murmuration videos showing enormous flocks of birds flying as if they were one; undulating, turning, responding as though they have a collective mind. Here's one is of a flock of starlings being hunted by a peregrine falcon one evening in Rome. The starlings fly so unpredictably that, at least this time, the falcon is unable to catch a meal.
Source: BBC One
According to the Cornell Lab of Ornithology, murmurations almost always arise in response to the detection of a predator; a falcon or a hawk that has come for its dinner, as the starlings in Rome. So, a bird or birds detect the predator and sound the alarm, which triggers the whole flock to take off. But, how do they stay together? Who decides where they're going next, and how does the rest of the flock get the message?
Young et al. report, in a 2013 paper in PLOS Computational Biology, that once in flight each bird is noticing and responding to the behavior only of its seven nearest neighbors. The murmuration, the movement of the group, then, is due to local responses that create the waves of motion that can be seen in the evening sky. There is no single leader, just many, many local responses happening almost simultaneously.
The same kinds of dynamics explain the movements of schools of fish as well. They work to some extent, but fish are routinely attacked by sharks, which can scoop up multiple individuals at a time, and surely sometimes birds of prey manage to snap up a luckless bird among the thousands or millions in a flock. But, most of the fish or the birds do get away, so it's a winning strategy for the group. Public Health in action.
Well-known, very prolific British epidemiologist George Davey Smith was interviewed on the BBC Radio 4 program The Life Scientific not long ago. He's a medical doctor with a degree in Public Health as well, so he's been trained to think in terms of both the population and the individual. He is currently interested in what genes can tell us about environmental influences on health. One of his contributions to this question is the analytical tool called Mendelian Randomization, which aims to tease out environmental triggers of a trait given a particular genetic risk factor. That is, the idea is to divide a study sample into individuals with and without a particular genetic variant, to determine whether their history of exposure to an apparent risk factor might be responsible for the disease. In this instance, the gene isn't modifiable, but exposure might be.
In the interview, Davey Smith said that his primary interest is in population health, and that if a Public Health measure can reduce incidence of disease, he's happy. So, if everyone in a population is on statins, say, and that reduces heart disease and stroke without major side effects, he would consider that a successful Public Health measure. Even if it's impossible to know just who's stroke or heart attack was prevented. Success of Public Health can only be evaluated on the population, not the individual level.
So much for personalized, predictive medicine. That's fine, my training is in Public Health, too, so I'm ok with that. Except that Davey Smith is also a fan of large, longitudinal studies maintained in perpetuity because, as he said, they have yielded more results at lower cost than most any other kind of epidemiological study.
But there are problems with such studies, and if the idea is to identify modifiable environmental risk factors, a major problem is that these studies are always retrospective. And, as we've written here so often, future environments are not predictable in principle. Presumably the aim of these large studies is to use Big Data to determine which Public Health measures are required to reduce risk of which diseases, and if that is done -- so that large segments of the population are put on statins or change from saturated to unsaturated fats or start to exercise or quit smoking -- this changes environmental exposures, and thus the suite of diseases that people are then at risk of.
So, Public Health has to always be playing catch up. Controlling infectious diseases can be said to have been a cause of the increase in cancer and obesity and heart disease and stroke, by increasing the number of people who avoided infectious disease to live to be at risk of these later diseases. So, in that sense, putting whole populations on statins is going to cause the next wave of diseases that will kill most of us, even if we don't yet know what these diseases will be. Maybe even infectious diseases we currently know nothing about.
Even though, after putting their favored Public Health measure into effect, all the starlings outwitted the falcon that particular night in Rome, they're all eventually going to die of something.
causation etiketine sahip kayıtlar gösteriliyor. Tüm kayıtları göster
causation etiketine sahip kayıtlar gösteriliyor. Tüm kayıtları göster
Somatic mutation beyond neurological traits. Part IV: the big mistake in genetics
The previous posts in this series were about the potential relevance of somatic mutation to neurologically relevant traits. I commented about ideas I've long had about the possible genetic etiology of epilepsies, but then about the more general relevance of somatic mutation for behavior and other less clinical traits, indeed, to positively beneficial traits. But the issues go much farther!
Fundamental units as the basis of science
Every science has fundamental units at the bottom of their causal scale, whose existence and properties can be assumed and tested, but below which we cannot go. The science is about the behavior or consequences of these units and their interactions. The fundamental unit's nature and existence per se are simply assumed. Physicists generally don't ask what is inside a photon or electron or neutron (or they say that these 'particles' are really 'waves'). In that sense, fundamental 'causes' are also defined but not internally probed. They don't really attempt to define 'force' except empirically or, for that matter, 'curved space-time'. You simply don't go there! Or, more precisely, if and when you venture into the innards of fundamental units, you do that by defining other even more fundamental units. When string theory tries to delve into unreal dimensions, they leave most other physicists, certainly the day-to-day ones behind. Generally, I think, physicists are usually more clear about this than biologists. The same in mathematics: we have fundamental axioms and the like that are accepted, not proven or tested.
Why is somatic mutation considered to be some sort of side-show in genetics?
What are biology's fundamental units? For historical reasons, evolutionary biology, which became much of the conceptual and theoretical foundation for biology, was about organisms. Before the molecular age, we simply didn't have the technology to think of organisms in the more detailed way we do now, but thought of them instead as a kind of unit in and of themselves.
Thus, the origins of ecology and phylogeny (before as well as after Darwin) were about whole organisms. Of course, it was long known that plants had leaves and animals had organs, and these and their structures and behavior (and pathologies) were studied in a way that was known to involve dissecting the system from its normal context. That is, organs were just integral parts of otherwise fundamental units. This was true even after microscopes were developed, Virchow and others had established the cell theory of life. Even after Pasteur and others began studying bacteria in detail, the bacterium itself was a fundamental unit.
But this was a major mistake. Dissecting organs to understand them did, when considered properly, allow the identification of digestion, circulation, muscle contraction, and the like. But the focus then, and still today in the genetic age, on the whole organism as a basically fundamental unit has had some unfortunate consequences. We know that genes in some senses 'cause' biological traits, but we treat an organism as a fundamental unit with a genotype, and that is where much trouble lies.
The cell theory made it obvious that you and I are not not just an indivisible fundamental unit, with a genotype as its fundamental characteristic. Theories of organisms, embryology, and evolution largely rest on that assumption, but it is a mistake, and somatic mutation is a major reason why.
The cell theory, or cell fact, really, makes it clear that you and I are clearly not indivisible causal units with a genotype. We know beyond dispute that cell division typically involves at least some DNA replication errors--'errors', that is, if you think life's purpose is to replicate faithfully. That itself is a bit strange, because life is an evolutionary phenomenon that is fundamentally about variation. Perhaps like most things in the physical world, the important issues have to do with the amount of variation.
The number of cell-divisions from conception through adulthood in humans is huge. It is comparable to the number of generations in a species, or even a species' lifespan. Modern humans have been around for, say, 100,000 generations (2 million years), far fewer than the number of cell divisions in a lifetime. In addition, the number of cells in a human body at any given time is in the many billions, and many or even most cells continue to renew throughout life. This is comparable to the species size of many organisms. The point is that the amount of somatically generated variation among cells in any given individual is comparable to the amount of germline variation in a species or even a species' history. And I have not included the ecological diversity of each individual organism, including the bacteria and viruses and other small organisms on, in, and through a larger organism.
By assuming that somatic mutational variation doesn't exist or is trivially unimportant--that is, by assuming that a whole organism is the fundamental unit of life, we are entirely ignoring this rich, variable, dynamic ecology. Somatic mutation is hard to study. There are many ways that a body can detect and rid itself of 'mutant' cells--that is, that differ from the parent cell at their bodily time and place. But to treat each person as if s/he has 'the' genotype of his/her initial zygote is a rash assumption or, perhaps a bit more charitably, a convenient approximation.
Oversimplification, deeper and deeper
In the same way that we can understand the moon's orbit around the earth by ignoring the innards of both bodies, so long as we don't care about small orbital details, we can understand an organism's life and relations to others including its kin, by ignoring the internal dynamics that life is actually mainly about. But much of what the whole organism is or does is determined by the aggregate of its nature and the distribution of its genotypes over its large collections of cells. We have been indulging in avoiding inconvenient facts for several decades now. Before any real reason to think or know much about somatic mutation (except, for example, rearrangements in adaptive the immune system), the grossness of approximation was at least more excusable. But those days should be gone.
Geneomewide mapping is one example, of course. It can find those things which, when inherited in the germline and hence present in all other cells (except where it's been mutated), affect particular traits. Typically, traits of interest are found by mapping studies to be affected by tens, hundreds, or even thousands of 'genes' (including transcribed RNAs, regulatory regions etc.). Each individual inherits one diploid genotype, unique to every person, and then around this is a largely randomly generated distribution of mutant cells. When hundreds of genes contribute, it just makes no sense to think that what you inherit is what you are.
It should also be noted that we have no real way even to identify the 'constitutive' genome of an organism like a person. We must use some tissue sample, like blood or a cheek swab. But those will contain somatic mutations that arose subsequent to conception. We basically don't look for them and indeed each cell in the sample will be different. Sequencing will generally identify the common nucleotide(s) at each site, and that generally will be the inherited one(s), but that doesn't adequately characterize the variation among the cells; indeed, I think it largely ignores it as technical error.
The roles and relevance of somatic mutation might be studiable in comparing large-bodied, long-lived species with small ones in which not many cell divisions occur. They might be predicted to be more accurately described by constitutive (inherited) genomes, than larger species. Likewise plants with diverse 'germ lines', such as the countless meristems in trees that generate seeds, compared to simpler plants, might be illuminating.
How to understand and deal with these realities, is not easy to suggest. But it is easy to say that for every plausible reason somatic mutation must have substantial effects on traits good, bad, and otherwise. And that means that we have been wrong to consider the individual to be a fundamental unit of life.
Fundamental units as the basis of science
Every science has fundamental units at the bottom of their causal scale, whose existence and properties can be assumed and tested, but below which we cannot go. The science is about the behavior or consequences of these units and their interactions. The fundamental unit's nature and existence per se are simply assumed. Physicists generally don't ask what is inside a photon or electron or neutron (or they say that these 'particles' are really 'waves'). In that sense, fundamental 'causes' are also defined but not internally probed. They don't really attempt to define 'force' except empirically or, for that matter, 'curved space-time'. You simply don't go there! Or, more precisely, if and when you venture into the innards of fundamental units, you do that by defining other even more fundamental units. When string theory tries to delve into unreal dimensions, they leave most other physicists, certainly the day-to-day ones behind. Generally, I think, physicists are usually more clear about this than biologists. The same in mathematics: we have fundamental axioms and the like that are accepted, not proven or tested.
Why is somatic mutation considered to be some sort of side-show in genetics?
What are biology's fundamental units? For historical reasons, evolutionary biology, which became much of the conceptual and theoretical foundation for biology, was about organisms. Before the molecular age, we simply didn't have the technology to think of organisms in the more detailed way we do now, but thought of them instead as a kind of unit in and of themselves.
Thus, the origins of ecology and phylogeny (before as well as after Darwin) were about whole organisms. Of course, it was long known that plants had leaves and animals had organs, and these and their structures and behavior (and pathologies) were studied in a way that was known to involve dissecting the system from its normal context. That is, organs were just integral parts of otherwise fundamental units. This was true even after microscopes were developed, Virchow and others had established the cell theory of life. Even after Pasteur and others began studying bacteria in detail, the bacterium itself was a fundamental unit.
 |
| Eukaryotic cell; figure from The Mermaid's Tale, Weiss and Buchanan, 2009 |
But this was a major mistake. Dissecting organs to understand them did, when considered properly, allow the identification of digestion, circulation, muscle contraction, and the like. But the focus then, and still today in the genetic age, on the whole organism as a basically fundamental unit has had some unfortunate consequences. We know that genes in some senses 'cause' biological traits, but we treat an organism as a fundamental unit with a genotype, and that is where much trouble lies.
The cell theory made it obvious that you and I are not not just an indivisible fundamental unit, with a genotype as its fundamental characteristic. Theories of organisms, embryology, and evolution largely rest on that assumption, but it is a mistake, and somatic mutation is a major reason why.
The cell theory, or cell fact, really, makes it clear that you and I are clearly not indivisible causal units with a genotype. We know beyond dispute that cell division typically involves at least some DNA replication errors--'errors', that is, if you think life's purpose is to replicate faithfully. That itself is a bit strange, because life is an evolutionary phenomenon that is fundamentally about variation. Perhaps like most things in the physical world, the important issues have to do with the amount of variation.
 |
| Mitotic spindle during cell division; from Wikipedia, Public Domain |
The number of cell-divisions from conception through adulthood in humans is huge. It is comparable to the number of generations in a species, or even a species' lifespan. Modern humans have been around for, say, 100,000 generations (2 million years), far fewer than the number of cell divisions in a lifetime. In addition, the number of cells in a human body at any given time is in the many billions, and many or even most cells continue to renew throughout life. This is comparable to the species size of many organisms. The point is that the amount of somatically generated variation among cells in any given individual is comparable to the amount of germline variation in a species or even a species' history. And I have not included the ecological diversity of each individual organism, including the bacteria and viruses and other small organisms on, in, and through a larger organism.
By assuming that somatic mutational variation doesn't exist or is trivially unimportant--that is, by assuming that a whole organism is the fundamental unit of life, we are entirely ignoring this rich, variable, dynamic ecology. Somatic mutation is hard to study. There are many ways that a body can detect and rid itself of 'mutant' cells--that is, that differ from the parent cell at their bodily time and place. But to treat each person as if s/he has 'the' genotype of his/her initial zygote is a rash assumption or, perhaps a bit more charitably, a convenient approximation.
Oversimplification, deeper and deeper
In the same way that we can understand the moon's orbit around the earth by ignoring the innards of both bodies, so long as we don't care about small orbital details, we can understand an organism's life and relations to others including its kin, by ignoring the internal dynamics that life is actually mainly about. But much of what the whole organism is or does is determined by the aggregate of its nature and the distribution of its genotypes over its large collections of cells. We have been indulging in avoiding inconvenient facts for several decades now. Before any real reason to think or know much about somatic mutation (except, for example, rearrangements in adaptive the immune system), the grossness of approximation was at least more excusable. But those days should be gone.
Geneomewide mapping is one example, of course. It can find those things which, when inherited in the germline and hence present in all other cells (except where it's been mutated), affect particular traits. Typically, traits of interest are found by mapping studies to be affected by tens, hundreds, or even thousands of 'genes' (including transcribed RNAs, regulatory regions etc.). Each individual inherits one diploid genotype, unique to every person, and then around this is a largely randomly generated distribution of mutant cells. When hundreds of genes contribute, it just makes no sense to think that what you inherit is what you are.
It should also be noted that we have no real way even to identify the 'constitutive' genome of an organism like a person. We must use some tissue sample, like blood or a cheek swab. But those will contain somatic mutations that arose subsequent to conception. We basically don't look for them and indeed each cell in the sample will be different. Sequencing will generally identify the common nucleotide(s) at each site, and that generally will be the inherited one(s), but that doesn't adequately characterize the variation among the cells; indeed, I think it largely ignores it as technical error.
The roles and relevance of somatic mutation might be studiable in comparing large-bodied, long-lived species with small ones in which not many cell divisions occur. They might be predicted to be more accurately described by constitutive (inherited) genomes, than larger species. Likewise plants with diverse 'germ lines', such as the countless meristems in trees that generate seeds, compared to simpler plants, might be illuminating.
How to understand and deal with these realities, is not easy to suggest. But it is easy to say that for every plausible reason somatic mutation must have substantial effects on traits good, bad, and otherwise. And that means that we have been wrong to consider the individual to be a fundamental unit of life.
Thoughts on the latest schizophrenia genetics report
The news and social media were headlining a report last week that presented some genetic findings, and even aspects of a possible causal mechanism, related to schizophrenia. As habitually skeptical readers of these daily stories, we wondered how substantial this claim is.
The report in question was a Nature paper by Sekar et al. that identifies variation in the very complex MHC genome region that, based on the authors' analysis, is statistically associated with schizophrenics relative to unaffected controls. These are variants in the number of copies of particular genes in the C4 'Complement' system. The authors show that gene copy number is correlated with gene expression level and, in turn, with some changes in brain tissue that may be related to functional effects in schizophrenia patients.
Comparing genotypes and disease status, in ~30,000 cases and controls of European ancestry, in 40 cohorts from 22 countries, the authors find that genotypes with higher C4 gene copy numbers are more frequent in schizophrenics, and there is a quantitative relationship between copy number and expression level in postmortem-tested neural tissue. The relevant potential mechanism involved may have to do with the pruning of synapses among neurons in the brain.
The authors estimate that the relative risk of the highest-copy number genotype is 1.27 times that of the lowest. The lowest risk genotype is rare in the population, comprising only about 7% of the sample population, meaning that almost everyone has a middling relative-risk genotype. That is comparable, say, to most of us having middling height or blood pressure. But the net population absolute risk of schizophrenia is about 1%, so that the absolute risks associated with these various genotypes are small and not even very different from each other. The careful work done by the authors has many different components that together consistently seem to show that these copy number differences do have real effects, even if the absolute risks are small.
How that effect or association arises is not clear, and the findings are certainly not the same as explaining schizophrenia as a C4 disease per se. As the authors note, around 100 or so other chromosome locations have been associated with the disease in genome-wide mapping studies that have been done. That means that if their results stand up to scrutiny, C4 variation is one component of what is basically a polygenic disorder. The association for each C4 genotype category is the effect averaged over all other contributing causes in those people. The absolute risk in individuals with a given copy number is still very small, and may depend on other genetic or environmental factors.
Schizophrenia is not a single disorder and has a spectrum of onset age, sex, symptoms, and severity of occurrence. Many authors have been warning against using a single term for this variety of traits. Whether that is relevant here or not remains to be seen, but at least as presented in the their paper, some of the current authors' results seem not to vary with age. This study doesn't address whether there is a smallish subset of individuals in each C4 category who are at much higher risk than the average for the category. However, the familial clustering of schizophrenia suggests this may be so, because family members share environments and also genomic backgrounds. One might expect that C4 genotypes are interacting with, or if not, being supplemented by, many other risk factors.
Even if average risk is not very high in absolute terms, this paper received the attention it did because it may be the first providing a seemingly strong case for a potentially relevant cellular mechanism to study, even if the specific effect on risk turns out to be quite small. It could provide a break in understanding the basic biology of schizophrenia, given the dearth of plausible mechanisms know so far.
Because the statistically riskier genotypes are found in a high percentage of Europeans, one would expect them to be found, if at varying frequencies, in other populations than Europeans. Whether their associated risks will be similar probably depends on how similarly the other risk factors are in other populations. C4 copy number variation must be evolutionarily old because there is so much of it, clearly not purged by natural selection--another indicator of a weak effect, especially because onset is often in the reproductive years and would seem to be potentially 'visible' to natural selection. So why is the C4 variation so frequent? Perhaps C4 provides some important neural function, and most variation causes little net harm, since schizophrenia is relatively rare at roughly 1% average risk. Or, copy number changes must happen regularly in this general MHC genome region, and can't effectively be purged, but is generally harmless. But there is another interesting aspect to this story.
The Complement system is within a large, cluster of genes generally involved in helping destroy invading pathogens that have been recognized. It is part of what is called the 'innate' immune system. Innate here means it does not vary adaptively in response to foreign bodies, like bacterial or viruses, that get into the blood stream. The adaptive immune system does that, and is highly variable for that reason; but once a foreigner is identified, the complement system takes part in destroying it. So it is curious that it would be involved in neural degeneration, unless it is responding to some foreign substance in the brain, or is an autoimmune reaction. But if the latter, how did it become so common? Or is the use of C4 genes in this neural context a pleiotropy--a 'borrowed' use of existing genes that arose for immunity-related functions but then came also to be used for a different function? Or is neural synapse regulation a kind of 'immune' function that hasn't been thought of in that way? Whatever it's doing, in modern society it contributes to problems about 1% of the time, for reasons for which this paper clearly will stimulate investigation.
Why does this system 'misfire' only about 1% of the time? One possible answer is that the C4 activity prunes synapse connections away normally in a random kind of way, but occasionally, by chance, prunes too much, leading to schizophrenia. The disease would in that sense be purely due to random bad luck, rather than interacting with other mechanisms or factors. The higher the copy number the more likely the bad luck but too weakly for selection to 'care'. However, that reason for the disease seems unlikely, for several reasons. First, mapping has identified about 100 or so genome regions statistically associate with schizophrenia risk, suggesting that the disease is not just bad luck. Secondly, schizophrenia is familial: close relatives seem to be at elevated risk, 10-fold in very close relatives and almost 50-fold in identical twins. This should not happen if the pathogenetic process is purely random, even though since haplotypes are inherited in close family members there could be a slight correlation in risk. Also, the authors cite several incidental facts that suggest that C4 plays some sort of systematic relevant functional role. But thirdly, since the absolute risk is so small, about 1%, one has to assume that C4 is not acting alone, but is directly interacting with, or is complemented by (so to speak) many other factors to which the unlucky victims have been exposed.
Something to test?
This might be a good situation in which to test a variant of an approach that British epidemiologist George Davey Smith has suggested as 'Mendelian randomization'. His idea is basically that, when there is a known candidate environmental risk factor and a known gene through which that environmental factor operates, one can compare people with a genetic variant exposed to an environmental risk factor to people with that genetic risk factor but not exposed to test whether the environmental factor really does affect risk.
Here, we could have a variant of that situation. We have the candidate gene system first, and could sort individuals having, say, the highest 'risk' genotypes, compared to the lowest, and see if any environmental or other systematic genomic differences are found that differentiates the two groups.
Interesting lead but not 'the' cause
Investigating even weakly causal factors could lead the way to discovering major pathogenic mechanisms or genetic or environmental contributors not yet known that interact with the identified gene region. There will be a flood of follow-up studies, one can be sure, but hopefully they will largely be focused investigations rather than repeat performances of association studies.
Given the absolute risks, which are small for given individuals, there may or may not be any reason to think that intervening on the C4 system itself would be a viable strategy even if it could be done. This still seems to be a polygenic--many-factorial--set of diseases, for which some other preventive strategy would be needed. Time will tell.
In any case, circumspection is in order. Remember traits like Alzheimer's disease, for which apoE, presenilins, beta-amyloid, and tau-protein associations were found years--or is it decades?--ago and still mystify to a great extent. Or the critical region of chromosome 21 in in Down syndrome that has, as far as we know, eluded intensive study for similarly long times. And there are many other similar stories related to what are essentially polygenic disorders with major environmental components. This one is, at least, an interesting one.
The report in question was a Nature paper by Sekar et al. that identifies variation in the very complex MHC genome region that, based on the authors' analysis, is statistically associated with schizophrenics relative to unaffected controls. These are variants in the number of copies of particular genes in the C4 'Complement' system. The authors show that gene copy number is correlated with gene expression level and, in turn, with some changes in brain tissue that may be related to functional effects in schizophrenia patients.
Comparing genotypes and disease status, in ~30,000 cases and controls of European ancestry, in 40 cohorts from 22 countries, the authors find that genotypes with higher C4 gene copy numbers are more frequent in schizophrenics, and there is a quantitative relationship between copy number and expression level in postmortem-tested neural tissue. The relevant potential mechanism involved may have to do with the pruning of synapses among neurons in the brain.
The authors estimate that the relative risk of the highest-copy number genotype is 1.27 times that of the lowest. The lowest risk genotype is rare in the population, comprising only about 7% of the sample population, meaning that almost everyone has a middling relative-risk genotype. That is comparable, say, to most of us having middling height or blood pressure. But the net population absolute risk of schizophrenia is about 1%, so that the absolute risks associated with these various genotypes are small and not even very different from each other. The careful work done by the authors has many different components that together consistently seem to show that these copy number differences do have real effects, even if the absolute risks are small.
How that effect or association arises is not clear, and the findings are certainly not the same as explaining schizophrenia as a C4 disease per se. As the authors note, around 100 or so other chromosome locations have been associated with the disease in genome-wide mapping studies that have been done. That means that if their results stand up to scrutiny, C4 variation is one component of what is basically a polygenic disorder. The association for each C4 genotype category is the effect averaged over all other contributing causes in those people. The absolute risk in individuals with a given copy number is still very small, and may depend on other genetic or environmental factors.
Schizophrenia is not a single disorder and has a spectrum of onset age, sex, symptoms, and severity of occurrence. Many authors have been warning against using a single term for this variety of traits. Whether that is relevant here or not remains to be seen, but at least as presented in the their paper, some of the current authors' results seem not to vary with age. This study doesn't address whether there is a smallish subset of individuals in each C4 category who are at much higher risk than the average for the category. However, the familial clustering of schizophrenia suggests this may be so, because family members share environments and also genomic backgrounds. One might expect that C4 genotypes are interacting with, or if not, being supplemented by, many other risk factors.
Even if average risk is not very high in absolute terms, this paper received the attention it did because it may be the first providing a seemingly strong case for a potentially relevant cellular mechanism to study, even if the specific effect on risk turns out to be quite small. It could provide a break in understanding the basic biology of schizophrenia, given the dearth of plausible mechanisms know so far.
Because the statistically riskier genotypes are found in a high percentage of Europeans, one would expect them to be found, if at varying frequencies, in other populations than Europeans. Whether their associated risks will be similar probably depends on how similarly the other risk factors are in other populations. C4 copy number variation must be evolutionarily old because there is so much of it, clearly not purged by natural selection--another indicator of a weak effect, especially because onset is often in the reproductive years and would seem to be potentially 'visible' to natural selection. So why is the C4 variation so frequent? Perhaps C4 provides some important neural function, and most variation causes little net harm, since schizophrenia is relatively rare at roughly 1% average risk. Or, copy number changes must happen regularly in this general MHC genome region, and can't effectively be purged, but is generally harmless. But there is another interesting aspect to this story.
The Complement system is within a large, cluster of genes generally involved in helping destroy invading pathogens that have been recognized. It is part of what is called the 'innate' immune system. Innate here means it does not vary adaptively in response to foreign bodies, like bacterial or viruses, that get into the blood stream. The adaptive immune system does that, and is highly variable for that reason; but once a foreigner is identified, the complement system takes part in destroying it. So it is curious that it would be involved in neural degeneration, unless it is responding to some foreign substance in the brain, or is an autoimmune reaction. But if the latter, how did it become so common? Or is the use of C4 genes in this neural context a pleiotropy--a 'borrowed' use of existing genes that arose for immunity-related functions but then came also to be used for a different function? Or is neural synapse regulation a kind of 'immune' function that hasn't been thought of in that way? Whatever it's doing, in modern society it contributes to problems about 1% of the time, for reasons for which this paper clearly will stimulate investigation.
Why does this system 'misfire' only about 1% of the time? One possible answer is that the C4 activity prunes synapse connections away normally in a random kind of way, but occasionally, by chance, prunes too much, leading to schizophrenia. The disease would in that sense be purely due to random bad luck, rather than interacting with other mechanisms or factors. The higher the copy number the more likely the bad luck but too weakly for selection to 'care'. However, that reason for the disease seems unlikely, for several reasons. First, mapping has identified about 100 or so genome regions statistically associate with schizophrenia risk, suggesting that the disease is not just bad luck. Secondly, schizophrenia is familial: close relatives seem to be at elevated risk, 10-fold in very close relatives and almost 50-fold in identical twins. This should not happen if the pathogenetic process is purely random, even though since haplotypes are inherited in close family members there could be a slight correlation in risk. Also, the authors cite several incidental facts that suggest that C4 plays some sort of systematic relevant functional role. But thirdly, since the absolute risk is so small, about 1%, one has to assume that C4 is not acting alone, but is directly interacting with, or is complemented by (so to speak) many other factors to which the unlucky victims have been exposed.
Something to test?
This might be a good situation in which to test a variant of an approach that British epidemiologist George Davey Smith has suggested as 'Mendelian randomization'. His idea is basically that, when there is a known candidate environmental risk factor and a known gene through which that environmental factor operates, one can compare people with a genetic variant exposed to an environmental risk factor to people with that genetic risk factor but not exposed to test whether the environmental factor really does affect risk.
Here, we could have a variant of that situation. We have the candidate gene system first, and could sort individuals having, say, the highest 'risk' genotypes, compared to the lowest, and see if any environmental or other systematic genomic differences are found that differentiates the two groups.
Investigating even weakly causal factors could lead the way to discovering major pathogenic mechanisms or genetic or environmental contributors not yet known that interact with the identified gene region. There will be a flood of follow-up studies, one can be sure, but hopefully they will largely be focused investigations rather than repeat performances of association studies.
Given the absolute risks, which are small for given individuals, there may or may not be any reason to think that intervening on the C4 system itself would be a viable strategy even if it could be done. This still seems to be a polygenic--many-factorial--set of diseases, for which some other preventive strategy would be needed. Time will tell.
In any case, circumspection is in order. Remember traits like Alzheimer's disease, for which apoE, presenilins, beta-amyloid, and tau-protein associations were found years--or is it decades?--ago and still mystify to a great extent. Or the critical region of chromosome 21 in in Down syndrome that has, as far as we know, eluded intensive study for similarly long times. And there are many other similar stories related to what are essentially polygenic disorders with major environmental components. This one is, at least, an interesting one.
Who should take statins? Is heart disease predictable?
Who should take statins.....besides everyone? I thought a lot about this when I was working on a lecture about predicting disease. The purpose of statins, of course, is to prevent atherosclerotic cardiovascular disease in people at risk (how well they do this is another issue). The challenge is to identify the people 'at risk'. I wrote about this in July, but I've been playing some more with the ideas and wanted to follow up.
Statins are a class of drug that, in theory, work by lowering LDL (low-denstity lipoprotein) levels. They do this by inhibiting HMG-CoA reductase, an enzyme that has a central role in the production of cholesterol in the liver. LDL, the so-called 'bad' cholesterol, isn't actually just cholesterol, but has been linked to risk of heart disease because, as a lipoprotein, its job is to transport cholesterol to and from cells. It is bound to cholesterol. What's measured when we have our blood drawn for a cholesterol test is LDL-C, the amount of cholesterol bound to LDL particles (LDL-C), as well as HDL-C, the 'good' cholesterol package, which transports LDL-C from cells, leading to lower blood cholesterol levels. Cholesterol makes plaque and plaque lines and hardens arteries, which occludes them and leads to stroke and heart attack. Lower the amount of LDL, and you lower the risk of arterial plaque deposits.
The connection between cholesterol and heart disease was first identified in the Framingham Study in the 1950's and 60's, and this lead directly to the search for drugs to lower cholesterol. Statins were developed in the 1970's and 80's, and after some fits and starts, began to be used in earnest in the late 1980's. Statins work by inhibiting the liver cells' synthesizing of new cholesterol, that is, cholesterol that isn't due taken in in the diet.
Akira Endo, one of the first scientists to look for cholesterol-lowering compounds, reviewed the history of statins in 2010. He described the many studies of the effects of these drugs, saying "The results in all these studies have been consistent: treatment with statins lowers plasma LDL levels by 25–35% and reduces the frequency of heart attacks by 25–30%" (Akira Endo, Proc Japan Acad, Series B, 2010).
A systematic review of the literature on the effectiveness of statins was published by the Cochrane Organization in 2012. The review reports, "Of 1000 people treated with a statin for five years, 18 would avoid a major CVD event which compares well with other treatments used for preventing cardiovascular disease." This suggests, of course, that 982 people took statins with no benefit, and perhaps some risk, as statins are associated with muscle pain, slightly increased risk of type 2 diabetes, liver damage, neurological effects, digestive problems, rash and flushing, and other effects. But more on this below.
So, who should take statins?
Until 2013, the recommendation was that anyone with a modest risk, as assessed by the Framingham Risk Calculator (I've read that that means from 6.5% to 10% 10-year risk) would likely be prescribed statins. The interesting thing, to me, about this risk calculator is that it's impossible to push the risk estimate past "greater than 30%", even at maximum allowable cholesterol, LDL, and systolic blood pressure, and being a smoker on blood pressure medication. Which means that there's a lot that this calculator can't tell us about our risk of CVD, based on the best risk factors known.
In 2013, the American Heart Association/American College of Cardiology revised their criteria for statins. Now, they are recommended for people who have had one CVD event in order to prevent another; for people with primary elevations of LDL-C greater than 190mg/dL; people 45-70 years old who have diabetes and LDL-C between 70 and 189mg/dL, and people 45-70 years old with LDL-C between 70 and 189mg/dL and estimated 10-year cardiovascular disease risk of 7.5% or higher.
The first three criteria are straightforward. If statins lower LDL, and lower LDL lowers risk of ASCVD (artherosclerotic cardiovascular disease), then taking them should be beneficial. But then we're back to a risk calculator again to estimate 10-year risk.
It has been revised. Now included are ethnicity (well, White, African American or other), and diabetic status (yes/no), and estimated lifetime risk. And, now it's possible to push 10-year risk up past 70%, which I discovered by playing around with the calculator a bit. Whether or not it's a more accurate predictor of a cardiovascular event is another question.
Here's the lowest risk I could come up with, 0.1% 10-year risk. The recommendations offered are not to prescribe statins.
Here's the highest risk I could force the calculator to estimate. Ten-year risk for a female with these risk factors is higher than for a male, but lifetime risk is lower. That seems strange, but ok, it must reflect association of risk factors including sex with disease at the population level.

Compared with the Framingham calculator, risk estimation seems to be getting more precise. Or at least bolder, with estimates up in the 70's. But is the new calculator actually better at predicting risk than the old one? A paper was recently published in JAMA addressing just this question ("Guideline-
Based Statin Eligibility, Coronary Artery Calcification, and Cardiovascular Events," Pursnani et al.) They identified 2435 people from the Framingham study who had never taken statins. Their medical history allowed the authors to determine that, based on the old guidelines, 14% would have been 'statin eligible' compared with 39%, based on the new 2013 guidelines.
Among those eligible by the old guidelines, 6.9% (24/348) developed CVD compared with 2.4% (50/2087) among noneligible participants (HR, 3.1; 95% CI, 1.9-5.0; P less than .001). Under the new guidelines, among those eligible for statins, 6.3% (59/941) developed incident CVD compared with only 1.0% (15/1494) among those not eligible (HR, 6.8; 95% CI, 3.8-11.9; P less than .001).
So, put a whole lot more people on statins, and you prevent an additional very small number of CVD events; 1.0% vs 2.4%. And, 93% of those ‘eligible’ for statins did not develop disease. Nor, of course, do statins prevent all disease. Actually, if everyone in the population were covered, statins would be preventing as many events as they could possibly prevent, but in a small minority of the population. That is, 90+% of people considered to be at 'high-risk' of disease don't go on to develop disease. Is it worth the side effects and cost to put so many more people on statins to prevent the 1.4% more CVD that these new guidelines are preventing? Well, heart disease is still the number one killer in rich countries, and 40+% of the population is currently taking statins, so a lot of people have decided that the benefits do outweigh the risks.
Another question, though, is more fundamental, and it concerns prediction. The calculator seems to now be predicting risk with some confidence. But, let's take a hypothetical person with a somewhat elevated risk. Her cholesterol is higher than the person above who's at lowest risk, but that's due to her HDL. Her systolic blood pressure is high at 180, which is apparently what bumps up her risk, but her 10-year risk is still not over 7.5% so the recommendation is not statins, but lifestyle and nutrition counseling. (Though, the definition of 'heart-healthy diet' keeps changing, so what to counsel this person with low risk seems a bit problematic, but ok.)
Now here's the same hypothetical person, but she's now a smoker, on medication to lower her blood pressure (and her b.p. is still high) and she has diabetes. Her 10-year risk of ASCVD jumps to 36.8%. This makes sense, given what we know about risk factors, right? The recommendation for her is high-intensity statins and lifestyle changes -- lose weight, do regular aerobic exercise, eat a heart-healthy diet, stop smoking (easy enough to say, so hard to do, which is another issue, of course, and the difficulty of changing all these behaviors is one reason that statins are so commonly prescribed).

But now I've lowered her total cholesterol by 70mg/dL, which is what statins ideally would do for her. Even so, the American College of Cardiology/American Heart Association recommendation is for 'high-intensity statin therapy' and lifestyle counseling. The calculator doesn't know this, but statins have already done everything they are likely to do for her.

So, let's add lifestyle changes. But, even when she quits smoking, her 10-year risk is 20%. So let's say we cure her diabetes -- even then, she's still at high enough risk (9%) that 'moderate to high-intensity statins' are recommended. I'm confused. I think even the calculator is confused. It seems there's a fuzzy area where statins are being recommended when what's left to do is, say, lower blood pressure, which statins won't do. This hypothetical woman probably needs to lower her weight to do that, and statins aren't going to help with that, either, but still they're recommended. Indeed, one of the criticisms of this risk calculator when it was released in 2013 was that it overestimates risk. Perhaps so, but it also seems to overestimate the benefit of statins.
Statins are a class of drug that, in theory, work by lowering LDL (low-denstity lipoprotein) levels. They do this by inhibiting HMG-CoA reductase, an enzyme that has a central role in the production of cholesterol in the liver. LDL, the so-called 'bad' cholesterol, isn't actually just cholesterol, but has been linked to risk of heart disease because, as a lipoprotein, its job is to transport cholesterol to and from cells. It is bound to cholesterol. What's measured when we have our blood drawn for a cholesterol test is LDL-C, the amount of cholesterol bound to LDL particles (LDL-C), as well as HDL-C, the 'good' cholesterol package, which transports LDL-C from cells, leading to lower blood cholesterol levels. Cholesterol makes plaque and plaque lines and hardens arteries, which occludes them and leads to stroke and heart attack. Lower the amount of LDL, and you lower the risk of arterial plaque deposits.
The connection between cholesterol and heart disease was first identified in the Framingham Study in the 1950's and 60's, and this lead directly to the search for drugs to lower cholesterol. Statins were developed in the 1970's and 80's, and after some fits and starts, began to be used in earnest in the late 1980's. Statins work by inhibiting the liver cells' synthesizing of new cholesterol, that is, cholesterol that isn't due taken in in the diet.
Akira Endo, one of the first scientists to look for cholesterol-lowering compounds, reviewed the history of statins in 2010. He described the many studies of the effects of these drugs, saying "The results in all these studies have been consistent: treatment with statins lowers plasma LDL levels by 25–35% and reduces the frequency of heart attacks by 25–30%" (Akira Endo, Proc Japan Acad, Series B, 2010).
A systematic review of the literature on the effectiveness of statins was published by the Cochrane Organization in 2012. The review reports, "Of 1000 people treated with a statin for five years, 18 would avoid a major CVD event which compares well with other treatments used for preventing cardiovascular disease." This suggests, of course, that 982 people took statins with no benefit, and perhaps some risk, as statins are associated with muscle pain, slightly increased risk of type 2 diabetes, liver damage, neurological effects, digestive problems, rash and flushing, and other effects. But more on this below.
So, who should take statins?
Until 2013, the recommendation was that anyone with a modest risk, as assessed by the Framingham Risk Calculator (I've read that that means from 6.5% to 10% 10-year risk) would likely be prescribed statins. The interesting thing, to me, about this risk calculator is that it's impossible to push the risk estimate past "greater than 30%", even at maximum allowable cholesterol, LDL, and systolic blood pressure, and being a smoker on blood pressure medication. Which means that there's a lot that this calculator can't tell us about our risk of CVD, based on the best risk factors known.
 |
| Framingham Risk Calculator |
In 2013, the American Heart Association/American College of Cardiology revised their criteria for statins. Now, they are recommended for people who have had one CVD event in order to prevent another; for people with primary elevations of LDL-C greater than 190mg/dL; people 45-70 years old who have diabetes and LDL-C between 70 and 189mg/dL, and people 45-70 years old with LDL-C between 70 and 189mg/dL and estimated 10-year cardiovascular disease risk of 7.5% or higher.
The first three criteria are straightforward. If statins lower LDL, and lower LDL lowers risk of ASCVD (artherosclerotic cardiovascular disease), then taking them should be beneficial. But then we're back to a risk calculator again to estimate 10-year risk.
 |
| ACC/AHA |
It has been revised. Now included are ethnicity (well, White, African American or other), and diabetic status (yes/no), and estimated lifetime risk. And, now it's possible to push 10-year risk up past 70%, which I discovered by playing around with the calculator a bit. Whether or not it's a more accurate predictor of a cardiovascular event is another question.
Here's the lowest risk I could come up with, 0.1% 10-year risk. The recommendations offered are not to prescribe statins.
 |
| Lowest 10-year risk |

Compared with the Framingham calculator, risk estimation seems to be getting more precise. Or at least bolder, with estimates up in the 70's. But is the new calculator actually better at predicting risk than the old one? A paper was recently published in JAMA addressing just this question ("Guideline-
Based Statin Eligibility, Coronary Artery Calcification, and Cardiovascular Events," Pursnani et al.) They identified 2435 people from the Framingham study who had never taken statins. Their medical history allowed the authors to determine that, based on the old guidelines, 14% would have been 'statin eligible' compared with 39%, based on the new 2013 guidelines.
Among those eligible by the old guidelines, 6.9% (24/348) developed CVD compared with 2.4% (50/2087) among noneligible participants (HR, 3.1; 95% CI, 1.9-5.0; P less than .001). Under the new guidelines, among those eligible for statins, 6.3% (59/941) developed incident CVD compared with only 1.0% (15/1494) among those not eligible (HR, 6.8; 95% CI, 3.8-11.9; P less than .001).
So, put a whole lot more people on statins, and you prevent an additional very small number of CVD events; 1.0% vs 2.4%. And, 93% of those ‘eligible’ for statins did not develop disease. Nor, of course, do statins prevent all disease. Actually, if everyone in the population were covered, statins would be preventing as many events as they could possibly prevent, but in a small minority of the population. That is, 90+% of people considered to be at 'high-risk' of disease don't go on to develop disease. Is it worth the side effects and cost to put so many more people on statins to prevent the 1.4% more CVD that these new guidelines are preventing? Well, heart disease is still the number one killer in rich countries, and 40+% of the population is currently taking statins, so a lot of people have decided that the benefits do outweigh the risks.
Another question, though, is more fundamental, and it concerns prediction. The calculator seems to now be predicting risk with some confidence. But, let's take a hypothetical person with a somewhat elevated risk. Her cholesterol is higher than the person above who's at lowest risk, but that's due to her HDL. Her systolic blood pressure is high at 180, which is apparently what bumps up her risk, but her 10-year risk is still not over 7.5% so the recommendation is not statins, but lifestyle and nutrition counseling. (Though, the definition of 'heart-healthy diet' keeps changing, so what to counsel this person with low risk seems a bit problematic, but ok.)
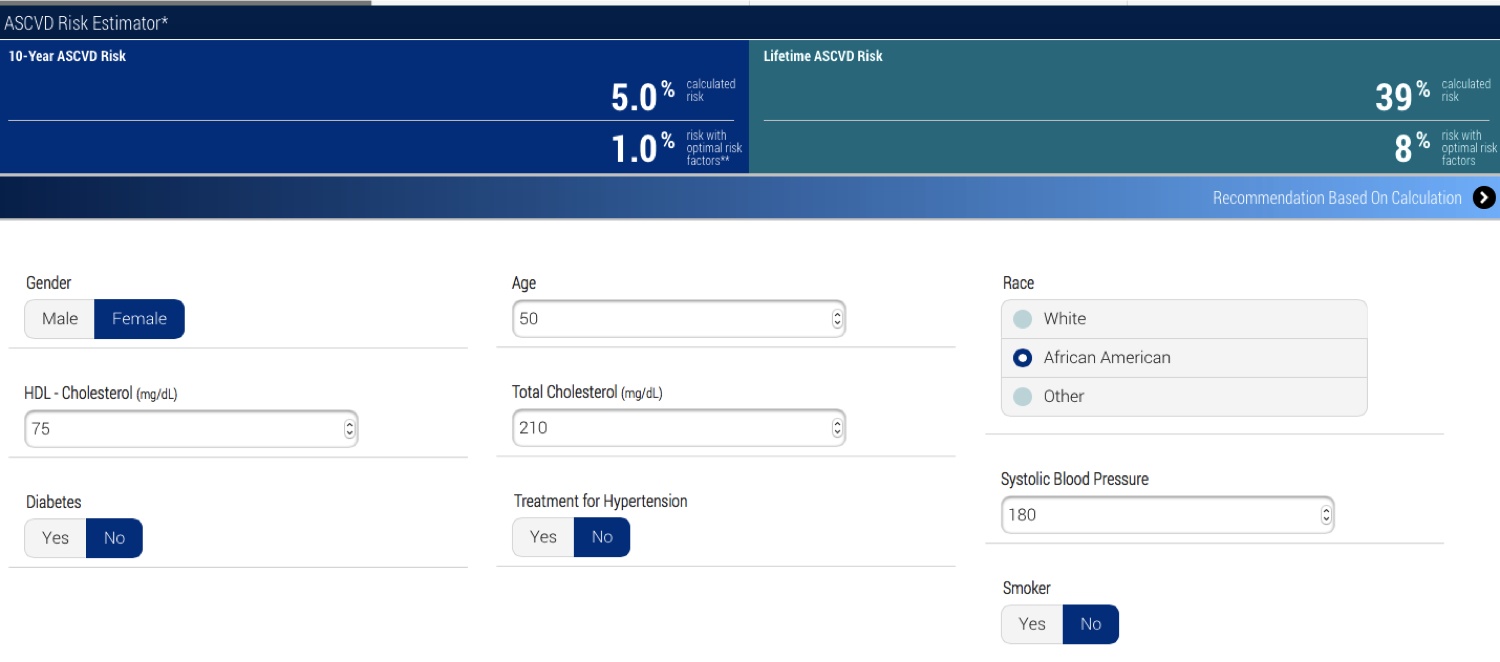 |
| Low enough risk that statins aren't advised. |
Now here's the same hypothetical person, but she's now a smoker, on medication to lower her blood pressure (and her b.p. is still high) and she has diabetes. Her 10-year risk of ASCVD jumps to 36.8%. This makes sense, given what we know about risk factors, right? The recommendation for her is high-intensity statins and lifestyle changes -- lose weight, do regular aerobic exercise, eat a heart-healthy diet, stop smoking (easy enough to say, so hard to do, which is another issue, of course, and the difficulty of changing all these behaviors is one reason that statins are so commonly prescribed).

But now I've lowered her total cholesterol by 70mg/dL, which is what statins ideally would do for her. Even so, the American College of Cardiology/American Heart Association recommendation is for 'high-intensity statin therapy' and lifestyle counseling. The calculator doesn't know this, but statins have already done everything they are likely to do for her.

So, let's add lifestyle changes. But, even when she quits smoking, her 10-year risk is 20%. So let's say we cure her diabetes -- even then, she's still at high enough risk (9%) that 'moderate to high-intensity statins' are recommended. I'm confused. I think even the calculator is confused. It seems there's a fuzzy area where statins are being recommended when what's left to do is, say, lower blood pressure, which statins won't do. This hypothetical woman probably needs to lower her weight to do that, and statins aren't going to help with that, either, but still they're recommended. Indeed, one of the criticisms of this risk calculator when it was released in 2013 was that it overestimates risk. Perhaps so, but it also seems to overestimate the benefit of statins.
Further, it seems there are a lot of type 1 errors here. That is, a lot of people are considered 'at-risk' who wouldn't actually develop cardiovascular disease. Risk of 7.5% means 7.5 of 100 people with a given, equal set of risk factors are expected to develop disease. That means that 92.5 would not. And that means that we have a pretty rough understanding of heart disease risk. The strongest risk factors we know -- smoking, high LDL-C, diabetes and hypertension -- can be expected to predict only a small fraction of events.
And that means that either something else is 'causing' cardiovascular disease in addition to these major known risk factors, or something is protecting people with these risk factors who don't go on to develop disease. Family history is a good or even the very best single predictor (why isn't it taken into account in these calculators?) which suggests that it's possible that genetic risk (or protection) is involved, but genome wide association studies haven't found genes with large effects. Of course, family history is highly conflated with environmental factors, too, so we shouldn't simply assume we need to look for genes when family history indicates risk. Anyway, it's unlikely that there are single genes responsible for ASCVD except in rare families, because that's the nature of complex diseases. Instead, many genes would be involved, but again as with most complex diseases, they would surely be interacting with environmental risk factors, and we don't yet know understand how to identify or really understand gene by environment interaction.
And then there's the truly wild card! All of these risks are based on the combinations of past exposures to measured lifestyle factors, but the mix of those and the rise of other new lifestyle factors, or the demise of past ones, means that the most fundamental of all predictors can itself not be predicted, not even in principle!
So, statins are a very broad brush, and a lot more people are being painted with them than in fact need to be. The problem is determining which people these are, but rather than zoom in with more precision, the updated calculator instead paints a whole lot more people with the brush. This isn't the calculator's fault. It's because understanding risk is difficult, ASCVD is a large and heterogeneous category, and prediction is very imprecise -- even for many 'simple' Mendelian disorders. If ASCVD were caused by a single gene, we'd say it had very low penetrance. And we'd want to understand the factors that affect its penetrance. That's the equivalent to where we are with cardiovascular disease.
I was interested to see that the 2013 ACC/AHA Guideline on the Assessment of Cardiovascular Risk says something that I have said so many times that I decided not to say it again in this post. But, I'm happy to see it elsewhere now. The guideline committee itself acknowledges the issue, so I'll let them explain the problem of assessing risk as their calculator does.
Finally, here's a meta-thought about all this. Ken and I were in Finland this month co-teaching a course, Logical Reasoning in Human Genetics, with colleagues, including Joe Terwilliger. Joe said multiple times, "We suck at finding candidate genes because we don't know anything about biology. We're infants learning to crawl." The same can be said about epidemiological risk factors for many complex diseases -- we suck at understanding the causes of these diseases, and thus we suck at prediction, because we don't really understand the biology.
And that means that either something else is 'causing' cardiovascular disease in addition to these major known risk factors, or something is protecting people with these risk factors who don't go on to develop disease. Family history is a good or even the very best single predictor (why isn't it taken into account in these calculators?) which suggests that it's possible that genetic risk (or protection) is involved, but genome wide association studies haven't found genes with large effects. Of course, family history is highly conflated with environmental factors, too, so we shouldn't simply assume we need to look for genes when family history indicates risk. Anyway, it's unlikely that there are single genes responsible for ASCVD except in rare families, because that's the nature of complex diseases. Instead, many genes would be involved, but again as with most complex diseases, they would surely be interacting with environmental risk factors, and we don't yet know understand how to identify or really understand gene by environment interaction.
And then there's the truly wild card! All of these risks are based on the combinations of past exposures to measured lifestyle factors, but the mix of those and the rise of other new lifestyle factors, or the demise of past ones, means that the most fundamental of all predictors can itself not be predicted, not even in principle!
So, statins are a very broad brush, and a lot more people are being painted with them than in fact need to be. The problem is determining which people these are, but rather than zoom in with more precision, the updated calculator instead paints a whole lot more people with the brush. This isn't the calculator's fault. It's because understanding risk is difficult, ASCVD is a large and heterogeneous category, and prediction is very imprecise -- even for many 'simple' Mendelian disorders. If ASCVD were caused by a single gene, we'd say it had very low penetrance. And we'd want to understand the factors that affect its penetrance. That's the equivalent to where we are with cardiovascular disease.
I was interested to see that the 2013 ACC/AHA Guideline on the Assessment of Cardiovascular Risk says something that I have said so many times that I decided not to say it again in this post. But, I'm happy to see it elsewhere now. The guideline committee itself acknowledges the issue, so I'll let them explain the problem of assessing risk as their calculator does.
By its nature, such an approach requires a platform for reliable quantitative estimation of absolute risk based on data from representative population samples. It is important to note that risk estimation is based on group averages, which are then applied to individual patients in practice. This process is admittedly imperfect; no one has 10% or 20% of a heart attack during a 10-year period. Individuals with the same estimated risk will either have or not have the event of interest, and only those patients who are destined to have an event can have their event prevented by therapy.It's the problem of using group data, which is all we've got, to make clinical decisions about individuals. It's the meta-analysis problem -- meta-analyses compile data from many individual studies to produce a single result that certainly reflects all the studies, because they were all included in the statistics, but it doesn't represent any of them with precision. Ultimately, it's the problem that these sorts of inferences must be based on statistical analysis of samples -- collections -- of individuals. We do not have an easy way around this, including the N of 1 studies currently being proposed.
Finally, here's a meta-thought about all this. Ken and I were in Finland this month co-teaching a course, Logical Reasoning in Human Genetics, with colleagues, including Joe Terwilliger. Joe said multiple times, "We suck at finding candidate genes because we don't know anything about biology. We're infants learning to crawl." The same can be said about epidemiological risk factors for many complex diseases -- we suck at understanding the causes of these diseases, and thus we suck at prediction, because we don't really understand the biology.
Kaydol:
Kayıtlar (Atom)
Rare Disease Day and the promises of personalized medicine
O ur daughter Ellen wrote the post that I republish below 3 years ago, and we've reposted it in commemoration of Rare Disease Day, Febru...
-
 Pakistan dizileri Hint dizilerinden farklı. Onlar gibi coşkulu olmuyor genelde. Bu yüzden yarım bıraktıklarım hayli fazla. Ama bu dizi ...
Pakistan dizileri Hint dizilerinden farklı. Onlar gibi coşkulu olmuyor genelde. Bu yüzden yarım bıraktıklarım hayli fazla. Ama bu dizi ... -
 O ur daughter Ellen wrote the post that I republish below 3 years ago, and we've reposted it in commemoration of Rare Disease Day, Febru...
O ur daughter Ellen wrote the post that I republish below 3 years ago, and we've reposted it in commemoration of Rare Disease Day, Febru... -
 W e discussed a Japanese pachinko machine in an earlier post , a pinball machine, as an example of the difference between randomness and det...
W e discussed a Japanese pachinko machine in an earlier post , a pinball machine, as an example of the difference between randomness and det...